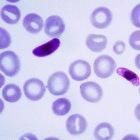
Fukushima
Fukushima
 HOSPITALISATION A DOMICILE / TAYSIR ASSISTA
HOSPITALISATION A DOMICILE / TAYSIR ASSISTA
Traitement de paludisme grave

TRAITEMENT ÉTIOLOGIQUE
Son but est la guérison radicale, c’est-à-dire la destruction de tous les parasites présents dans l’organisme.
Seuls les schizonticides, actifs sur les formes asexuées intraérythrocytaires, sont utilisés. La voie intraveineuse est impérative afin d’obtenir le plus rapidement possible une concentration plasmatique efficace (fig 3).
Sa difficulté actuelle réside dans la multiplication des souches chimiorésistantes, imposant l’utilisation d’un schizonticide très puissant pendant une durée prolongée [89].
Quinine
Alcaloïde extrait du quinquina, la quinine demeure l’antipaludique de référence.
Présentation
On emploie, en thérapeutique, des sels basiques (chlorhydrate ou formiate) dont la teneur en quinine-base doit être connue, et des mélanges d’alcaloïdes (tableau II). La posologie est exprimée en quinine-base quand la spécialité pharmaceutique contient uniquement un sel de quinine (Quinoformet, dichlorhydrate de quinine) ou en alcaloïde-base quand la quinine est associée à d’autres alcaloïdes actifs (Quinimaxt) (tableau III).
Protocole
Classiquement, la posologie de quinine était de 24 mg/kg/24 h de quinine-base. Mais la chloroquinorésistance de certaines souches de P. falciparum, responsable d’une diminution de sensibilité à la quinine, en particulier en Thaïlande où les souches multirésistantes sont de plus en plus nombreuses [65], a conduit certains auteurs du Sud-Est asiatique à proposer l’utilisation de doses de charge de 17 mg/kg de quinine-base suivies de réinjections, afin d’obtenir plus rapidement des taux sanguins efficaces, le traitement étant poursuivi pendant 7 jours par voie intraveineuse selon le protocole classique [90].
En Afrique, bien que la sensibilité des souches de P. falciparum à la quinine soit plus importante que celle des souches thaïlandaises [93], on a pu montrer, chez l’adulte semi-immun, sans que son intérêt en terme de mortalité ait été apprécié, que l’utilisation de doses de charge diminuait la durée du coma. Chez l’enfant, l’intérêt de doses de charge est contesté en raison de concentrations sanguines supérieures de 43 % à celles de l’adulte traité avec le même protocole et donc d’un risque toxique plus important [93].
La bonne tolérance clinique de la quinine et le risque potentiel d’un retard d’efficacité dans le traitement étiologique d’un paludisme grave ont conduit la conférence de consensus consacrée à la prise en charge et à la prévention du paludisme d’importation à P. falciparum à préconiser l’utilisation d’une dose de charge de quinine chez tout sujet non immun présentant un paludisme grave [63].
En pratique, la dose de charge, 17 mg/kg de quinine-base (20 mg/kg de Quinoformet ou de dichlorhydrate de quinine), ou 17 mg/kg d’alcaloïde-base (17 mg/kg de Quinimaxt) sera administrée dans du sérum glucosé à 5 % en 4 heures, suivie d’un traitement d’entretien de 8 mg/kg toutes les 8 heures en perfusions de quatre heures ou de 24 mg/kg/24 h en perfusion continue au pousse-seringue électrique [63].
La quininémie est dosée à la quatrième heure, puis quotidiennement ; elle doit se situer entre 10 et 15 mg/L (30-35 mmol/L).
En cas d’insuffisance rénale, la posologie n’est pas modifiée pendant les 48 premières heures, puis réduite d’environ un tiers [6]. Chez le patient en hémodialyse [77] ou en hémofiltration [20], la demi-vie n’est pas allongée et les posologies ne doivent pas être modifiées.
En revanche, en cas d’insuffisance hépatique, il faut diminuer la dose de moitié dès la deuxième perfusion, la quinine étant métabolisée à 80 % par le foie [6].
Dès que la parasitémie a disparu et que la voie orale est possible, en général au bout de 2 à 4 jours, le relais est pris par la quinine orale jusqu’au septième jour. En cas de paludisme d’importation, une chimioprophylaxie ultérieure est inutile.
Adjuvants et alternatives à la quinine
Antibiotiques : doxycycline, clindamycine
Plusieurs antibiotiques ont une activité antiplasmodiale sur les modèles expérimentaux ou sur culture in vitro de P. falciparum multirésistant [2].
Parmi les nombreux produits testés, seules la doxycycline (Vibraveineuset, 100 mg par voie intraveineuse toutes les 12 h, indication hors autorisation de mise sur le marché [AMM]) et en cas de contre-indication aux cyclines, la clindamycine (Dalacinet, 10 mg/kg par voie intraveineuse toutes les 8 heures, indication hors AMM) sont actuellement proposées au cours du paludisme grave comme traitement adjuvant et synergique en cas de sensibilité diminuée à la quinine, en particulier chez des patients provenant de zones particulières de l’Asie du Sud-Est (zones frontalières forestières Thaïlande-Cambodge et Thaïlande-Myanmar) ou de la forêt amazonienne [63].La durée du traitement est de 7 jours pour la doxycycline, de 5 jours pour la clindamycine. Il est contre-indiqué chez l’enfant de moins de 8 ans et la femme enceinte.
Artémether [32, 91]
Bien que la quinine reste efficace dans le traitement du paludisme multirésistant, cette efficacité semble décliner même avec l’utilisation de doses de charge. Par ailleurs, des souches résistantes à la quinine bilieuse hémoglobinurique. Ces différents éléments ont conduit les chercheurs à s’intéresser à de nouvelles molécules, comme les dérivés du qinghaosu, préparées à partir de feuilles d’une armoise connue en Chine pour son efficacité sur les fièvres. Ces dérivés sont au nombre de trois : l’artémisinine utilisée par voie orale en Extrême-Orient pour le traitement curatif du paludisme bénin à P. falciparum ou P. vivax, l’artésunate et l’artémether.
L’artémether a été utilisé par voie intramusculaire avec une dose de charge de 3,2 mg/kg, puis une dose d’entretien de 1,6 mg/kg toutes les 12 à 24 heures. Les injections ne sont pas douloureuses. Aucun effet toxique local ou général n’a été noté chez l’homme, alors qu’une neurotoxicité a été démontrée expérimentalement. Depuis 1982, les cinq études qui ont été publiées ont observé une clairance d’élimination parasitaire moyenne de 43 heures.
En France, l’artémether (Paluthert) n’a pas l’AMM. Il est utilisé grâce à une autorisation temporaire d’utilisation nominative en traitement curatif des formes résistantes à la quinine (persistance d’une parasitémie avec formes asexuées après 3 jours de traitement bien conduit par la quinine ou réapparition d’une parasitémie dans les 7 jours suivant la fin du traitement de 5 à 7 jours) ou en cas de paludisme grave contracté dans une zone de résistance à la quinine (Asie du Sud-Est en particulier). Il est également indiqué chez les patients présentant une fièvre bilieuse hémoglobinurique ou une autre contre-indication formelle à la quinine [22].
Chez l’adulte, le schéma thérapeutique actuel comprend une injection intramusculaire de 80 mg, deux fois par jour le premier jour, puis 1,6 mg/kg en une seule injection intramusculaire les 4 jours suivants.
Autres antipaludiques
Ils n’ont pratiquement plus aucune place dans le traitement du paludisme grave.
La quinidine, autre alcaloïde du quinquina, aussi efficace que la quinine, est utilisée en Thaïlande en cas de diminution de sensibilité à la quinine [90]. Elle s’emploie par voie intraveineuse en perfusion continue sous forme de gluconate de quinidine à la posologie de 30-35 mg/kg/24 h après une dose de charge de 10 mg/kg en 2 heures. Les effets cardiaques restent modérés si l’on respecte ce protocole.
La chloroquine (Nivaquinet), amino-4-quinoléine de synthèse, est surtout utilisée par les Anglo-Saxons par voie intramusculaire à la posologie de 25 mg/kg/24 h. Le risque cardiaque étant relativement important, des morts subites ont été rapportées chez l’enfant de moins de 3 ans [95], et la multiplication des souches chloroquinorésistantes en Afrique noire expliquent que son utilisation soit de plus en plus remise en question dans le cadre du paludisme grave.La méfloquine (Lariamt) et l’halofantrine (Halfant), aminoalcaloïdes utilisables uniquement par voie entérale, ont pu être proposés à la posologie de 25 mg/kg en trois prises, en cas de résistance avérée à la quinine [95].
Autres thérapeutiques curatives
Exsanguinotransfusion
Introduite par Gyr et al [30] en 1974, elle a pour but de faire baisser rapidement la parasitémie, d’épurer les débris cellulaires résultant de la schizogonie et les différents médiateurs plasmatiques impliqués dans la physiopathologie du paludisme grave [71].
L’exsanguinotransfusion permettrait également une correction rapide de l’anémie et des troubles de la coagulation [71].
Ses indications restent discutées. La densité parasitaire (supérieure à 10 % en règle générale) proposée par certains auteurs [52] ne peut être retenue car il ne s’agit pas d’un critère absolu de gravité [95], en particulier en cas de souche chloroquinorésistante. L’absence d’évolution favorable après 36 heures de traitement bien conduit apparaît, en revanche, plus pertinente [71].
Plusieurs observations isolées font état d’une amélioration spectaculaire de l’état de conscience [71], alors que d’autres rapportent des complications mortelles [52].
Au total, l’absence d’étude randomisée rend très difficile l’évaluation de l’efficacité de l’exsanguinotransfusion dans le paludisme grave.
En pratique, son utilisation n’est pas recommandée par la conférence de consensus consacrée au paludisme d’importation [63].
Antimédiateurs
Le rôle des cytokines, essentiellement le TNF, dans la physiopathologie du paludisme grave, explique les tentatives d’utilisation d’anticorps monoclonaux anticytokines qui ont permis de raccourcir la durée de la fièvre, mais sans modification de la clairance parasitaire et de la mortalité [89].
TRAITEMENT SYMPTOMATIQUE
Tout patient présentant un paludisme grave doit être hospitalisé en soins intensifs en raison du risque permanent de complication brutale mettant en jeu le pronostic vital et nécessitant la mise en oeuvre de techniques de réanimation.
Traitement des convulsions
Les convulsions sont contrôlées dans la plupart des cas par le diazépam (Valiumt) 10 mg intraveineux chez l’adulte, 0,15 mg/kg chez l’enfant, à renouveler éventuellement.Le phénobarbital (Gardénalt), moins actif, est cependant plusmaniable car moins dépresseur respiratoire. Il peut s’administrer par voie intramusculaire : 100 à 150 mg chez l’adulte, 1 mg/kg chez l’enfant. La prévention des convulsions chez l’enfant peut être obtenue par une injection unique de phénobarbital à la posologie de 3,5 mg/kg.
Autres mesures
Une oxygénothérapie est toujours utile comme lors de tout état infectieux sévère.
La ventilation artificielle est parfois indispensable : coma avec score de Glasgow inférieur ou égal à 8 ou score de Molyneux inférieur à 2, dépression respiratoire secondaire à l’utilisation d’anticonvulsivants.
Un remplissage vasculaire est pratiquement toujours indiqué, en raison de la constance de l’hypovolémie [70]. Il est réalisé sous surveillance hémodynamique de la pression veineuse centrale, ou mieux de la pression artérielle pulmonaire d’occlusion.
L’utilisation d’amines vasoactives est, en revanche, rarement utile en dehors des formes avec choc hyperkinétique avéré [69].
L’épuration extrarénale, hémodialyse [77], hémofiltration continue [34], peut s’avérer indispensable en cas d’insuffisance rénale aiguë.La transfusion de concentrés érythrocytaires pour obtenir une concentration d’hémoglobine supérieure à 10 g/dL est fondamentale pour améliorer le transport d’oxygène.
THÉRAPEUTIQUES DISCUTÉES ET DANGEREUSES
Corticoïdes
Utilisés dans le but de diminuer l’oedème cérébral, ils sont actuellement abandonnés [63].En effet, ils ne diminuent pas la mortalité et sont responsables d’une augmentation significative des complications,
en particulier des pneumopathies et des hémorragies digestives [86].
Héparine et anti-inflammatoires non stéroïdiens
Chez l’animal, l’héparine réduit la parasitémie, la thrombopénie, les altérations de l’hémostase et la mortalité. Par ailleurs, les antiinflammatoires non stéroïdiens, bloquant les cyclo-oxygénases, diminuent la fréquence des complications dues au TNF. Chez l’homme, ces deux classes de médicaments ne modifient pas l’évolution du paludisme.
De la même façon, l’intérêt de l’héparine dans le traitement de la CIVD est de plus en plus contesté [95]. En revanche, une prévention de la maladie thromboembolique par l’héparine standard ou l’héparine de bas poids moléculaire est indispensable chez l’adulte [70].
Plasma frais congelé
Il est réservé, ainsi que l’antithrombine III, aux cas rares s’accompagnant d’une CIVD avérée.
Antibiothérapie systématique
Elle n’a jamais fait la preuve de son efficacité, malgré l’immunodépression profonde qui accompagne tout paludisme grave et qui est responsable de surinfections bactériennes
redoutables.
Il faut cependant insister sur la nécessité d’une asepsie rigoureuse dans tous les gestes de réanimation, et tout particulièrement chez le patient ventilé pour lequel le risque de pneumopathie nosocomiale est majeur [70].
| Tableau II. – Présentations commerciales de la quinine par voie parentérale. | |||
| Spécialité | Laboratoire | Sel de quinine | Présentation-composition |
| Quinoforme | Synthélabo | Formiate de quinine | Solution injectable Ampoules de 2 mL contenant 500 mg de sel Quinine-base : 87,6 % |
| Quinimax | Sanofi Winthrop | Gluconate de quinine et de quinidine Chlorhydrate de cinchonine et de cinchonidine | Solution injectable Ampoules de 1, 2, 4 mL contenant par mL : - gluconate de quinine : 192,56 mg - gluconate de quinidine : 5,29 mg - chlorhydrate de cinchonine : 1,06 mg - chlorhydrate de cinchonidine : 1,00 mg Quinine-base : 62,3 % (125 mg d’alcaloïdes-base/mL) |
| Dichlorhydrate de quinine | Pharmacie centrale des Hôpitaux | Dichlorhydrate de quinine | Solution injectable Ampoules de 10 mL contenant 100 ou 300 mg de sel Quinine-base : 81,7 % |